Prediction of Biological Activity Spectra for Substances:
in House Applications and Internet Feasibility
Vladimir Poroikov, Dmitrii Filimonov, Tatyana
Gloriozova,
Alexei Lagunin, Alla Stepanchikova
Institute of Biomedical Chemistry RAMS, Pogodinskaya Str., 10, 119832,
Moscow, Russia;
Tel: +7 095 246-3380; Fax: +7 095 245-0857; E-mail: vvp@ibmh.msk.su
Abstract: Computer prediction of biological activity spectra for drug-like organic substances is presented. New leads found on the basis of predicted biological activity spectra are considered. Possibilities for in house and Internet applications of this approach in computer-aided drug discovery are discussed.
Keywords: biological activity spectrum, computer-aided prediction, computer system PASS (Prediction of Activity Spectra for Substances), applications in computer-aided drug discovery, prediction via Internet.
Introduction
Methods
PASS
Elements
The
Training Set.
Chemical
Structure Description.
Biological
Activity Description.
Mathematical
Approach.
Quality of
Prediction
Interpretation
of the Prediction's Results
Various Applications of PASS
Revealing
New Effects and Mechanisms of Action
Finding
the Most Probable New Leads with Required Activity Spectra
Selecting
the Most Prospective Compounds for High Throughput Screening
Determining
the Screens that are More Relevant for a Particular Compound
Examples of PASS Use
Biological
Activity Spectrum Prediction via Internet
Conclusions
Acknowledgments
Tables
References
Most of known biologically active substances have many different biological activities that cause both main (therapeutic) and supplementary (side) actions. Some of these activities are found during the initial preclinical study of lead compound; some others are found in clinical trials of drug-candidate (see, for example, the Fluorouracil's activities in Table 1). Sometimes additional activities are discovered in many years after the first launching of a drug that becomes the base for a new therapeutic application (see some examples in Table 2).
Most of computer-aided methods used in the process of drug-discovery are applied to a single activity of studied compounds belong the same chemical series [1-5]. These methods are used for finding and optimization of new leads consequently taking into account the required activity, pharmacokinetic properties, etc.
Computer approach to predicting simultaneously the pharmacological effects, mechanisms of action and specific toxicities on the basis of compound's structural formula might provide the supplementary information to select the most prospective leads from the set of compounds under study. An idea about such computer-aided approach was proposed by Victor Avidon more than twenty years ago [6, 7]. This technology has been formerly developed and tested in the framework of National registration system of new chemical compounds synthesized in the USSR [8, 9]. Further it has been essentially reconsidered several times taking into account both theoretical analysis of the methods and accumulated experience of its application in finding new leads [10-14]. Current version of computer system PASS predicts 408 pharmacological effects, mechanisms of action, mutagenicity, carcinogenicity, teratogenicity & embryotoxicity [15] (see also: http://www.ibmh.msk.su/PASS/default.htm).
Here we present the approach used in computer system PASS (Prediction of Activity Spectrum for Substance) that predicts the biological activity of chemical compounds, examples of practical application of this approach in finding new leads, and new feasibility that provides the prediction of biological activity spectrum for a compound via Internet.
The approach used in PASS is based on the suggestion that Activity = function (Structure). Thus, by "comparing" the structure of a new compound with structures of well-known biologically active substances it is possible to estimate if a new compound may have a particular effect. Actually, the mental work of medicinal chemists is based on this principle. However, computer system PASS operates with many thousands of substances from the training set, and provides more objective estimate for probability to find any activity in a compound comparing to any researcher.
The process of PASS development and use is shown in Figure 1.
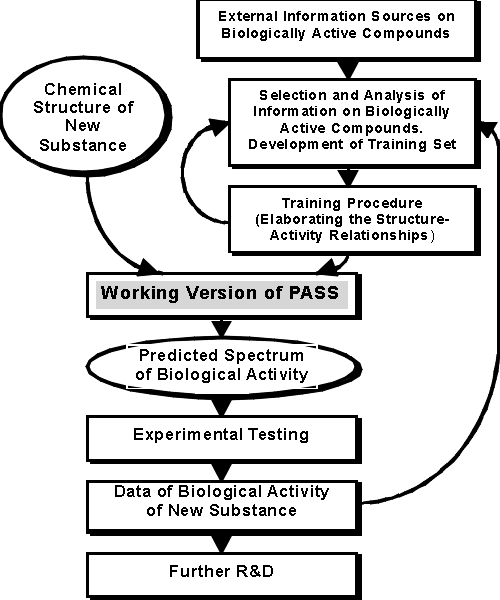
Figure 1. The Process of PASS Development
Principal elements of computer system PASS include the Training Set, Chemical Structure Description, Biological Activity Description, and the Mathematical Approach. They are described in more details below.
PASS-C 4.00 training set consists of about 28,000 of biologically active compounds, from which about 15,000 substances are already launched drugs, and about 13,000 drug-candidates are under the clinical or advanced preclinical testing now. Since 1972 this training set is compiled from many sources including: open publications, patents, databases, "gray" literature, etc. For the majority of compounds included into the training set special literature's search has been carried out to characterize the experimentally determined biological activity spectrum of each compound in details.
Structure of the compound is described by Multilevel Neighborhoods of Atoms (MNA) descriptors in computer system PASS. More detailed presentation of these descriptors is prepared as the separate publication. One example of description for the compound with structural formula H3C-CH2-OH, reflected the general idea, is given below.
At the first step, the 1st and 2nd levels neighborhoods for atoms are generated:
0 |
1 | 2 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
At the second step, the atoms- neighborhoods in MNA descriptors, that occurred in the molecule for several times, are reduced to the different once-occurred descriptors:
H(C(HHHC))
H(C(HHCO))
H(O(HC))
C(H(C)H(C)H(C)C(HHCO))
C(H(C)H(C)C(HHHC)O(HC))
O(H(O)C(HHCO))
Despite the possibility to continue the procedure calculating the 3rd, 4th, etc. levels of atoms' neighborhoods, only descriptors of the 1st and 2nd levels are used, because this approximation is shown to provide the best quality of prediction.
Biological activity is the result of chemical compound's interaction with biological entity. In clinical study biological entity is represented by human organism. In preclinical testing it is the experimental animals (in vivo) and experimental models (in vitro). Biological activity depends on peculiarities of compound (structure and physico-chemical properties), biological entity (species, sex, age, etc.), mode of treatment (dose, route, etc.).
Any biologically active compound reveals wide spectrum of different effects. Some of them are useful in treatment of definite diseases but the others cause various side and toxic effects. Total complex of activities caused by the compound in biological entities is called the "biological activity spectrum of the substance".
Biological activity spectrum of a compound presents every its activity despite of the difference in essential conditions of its experimental determination. If the difference in species, sex, age, dose, route, etc. is neglected the biological activity can be identified only qualitatively (yes/none). Thus, "biological activity spectrum" is defined as the "intrinsic" property of compound depending only on its structure and physico-chemical characteristics.
PASS-C 4.00 covers 408 kinds of biological activities included basic pharmacological effects, action mechanisms and specific toxicities which are presented in the corresponding Activity List (http://www.ibmh.msk.su/PASS/descript33.htm). Here, No. is the number of activity; Act.N. is the number of compounds that have the particular activity in the training set; MEP is the maximal error of prediction in percentage estimated in leave-one-out cross validation procedure.
Accuracy and efficiency of more than 200 various mathematical approaches were tested to select the most relevant algorithms [16]. One of the methods that provides satisfactory quality of prediction is described below in more details.
Designations:
n is the total amount of compounds in the training set;
ni is the amount of compounds, that have the descriptor i;
nj is the amount of compounds, that reveal the activity j;
nij is the amount of compounds, that have both the descriptor i and the activity
j;
pj = nj/n is the estimate of a priori probability of activity
j;
pij = nij/ni is the estimate of the conditional probability of the activity j
for the descriptor i;
m is the number of descriptors for the compound under prediction;
ri = ni/(ni + 0.5/m) is the regulating factor;
Prj is the initial estimate of the probability of the
activity j for the compound under prediction;
CPj is the cutting point;
E1j(CPj) is the estimate of 1st kind error probability;
E2j(CPj) is the estimate of 2nd kind error probability;
The 1st kind error is observed when the compound under
prediction actually is active but Prj < CPj;
The 2nd kind error is observed when the compound under prediction is considered
as inactive but Prj > CPj;
LOO is the leave-one-out procedure:
for each compound in the training set the values n, ni, nj, nij are changed to n-1, ni-1, and nj-1, nij-1 when it has activity j, and the estimates Prj are calculated.
MEP is the maximal error of prediction (see below).
Algorithm of Prediction:
For the compound under prediction structural descriptors are generated. For each activity the following values are calculated:
uj = SiArcSin{ri(2pij-1)}, vj = SiArcSin{ri(2pj-1)}
sj = Sin(uj/m), tj = Sin(vj/m)
Prj = (1+(sj-tj)/(1-sjtj))/2
Validation criterion:
For each compound in the training set the LOO estimates of
Prj are calculated.
For each activity the estimates of E1j(CPj) and E2j(CPj) are calculated. The
cutting points CPj* which provides equality:
E1j(CPj*) = E2j(CPj*)
are calculated. The maximal error of prediction MEP is:
MEPj = E1j(CPj*) = E2j(CPj*)
Results of Prediction:
The probability to be active is:
Pa = E1j(Prj)
The probability to be inactive is:
Pi = E2j(Prj)
The result of prediction is presented as the list of activities with appropriate Pa and Pi, sorted in descending order of the difference (Pa-Pi)>0.
In case when (1) the probabilities for 408 different activities are estimated simultaneously, and (2) the ideal training set should include all referenced biologically active compounds from literature, the best estimate of prediction's quality can be calculated by leave-one-out cross validation. Each of the compounds is subsequently removed from the training set and the prediction of its activity spectrum is carried out on the basis of the remaining part of the training set. The result is compared to the known activity of a compound, and the maximal error of prediction (MEP) is calculated and averaged
through the all compounds and activities.
For PASS-C 4.00 it is shown that this error is about 0.15, thus the average accuracy of prediction in LOO cross-validation is about 0.85. Such accuracy is enough for practical use of the system PASS, especially taking into account that the expected frequency of random guess-work in case of 408 activities is 1/408 » 0.0025.
Complete list of MEPs for each of 408 activities is given in the List of Activities (http://www.ibmh.msk.su/PASS/descript33.htm).
In the "Results of Prediction" one obtain the total number of MNA descriptions in her compound, and the number of descriptors which are new in comparison with the descriptors in 28,000 compounds from the PASS-C 4.00 training set. If the number of new descriptors is more than 3, the result of prediction may be not reliable.
In the predicted biological activity spectrum for the compound Pa and Pi are the estimates of probability to be active and inactive respectively. Their values vary from 0.000 to 1.000. Only activities with Pa > Pi are considered as possible for a particular compound.
If Pa > 0.7 the chance to find the activity in experiment is high, but in many cases the compound may occur to be the close analogue of known pharmaceutical agents.
If 0.5 < Pa < 0.7 the chance to find the activity in experiment is less, but the compound is not so similar to known pharmaceutical agents.
If Pa < 0.5 the chance to find the activity in experiment is even more less, but if it will be found the compound might occur to be a New Chemical Entity.
Thus, one may choose which activities have to be tested in her compounds on the basis of compromise between expected novelty of pharmacological agent and risk to obtain the negative result in experimental testing.
Certainly, the researcher will also take into account the particular interest to some kinds of activity, experimental facilities, etc.
As described above, computer system PASS-C 4.00 predicts simultaneously 408 kinds of activity with mean accuracy of prediction about 85% (leave-one-out cross validation) on the basis of the only compound's structural formula. It means that the system can be applied to compounds either synthesized or only planned to be synthesized.
Taking into account that the calculation of biological activity spectra for 1000 compounds in ordinary IBM PC Pentium/120 MHz takes about 5 minutes, one can effectively use PASS-C for predicting activity spectra of many compounds from large in-house and commercial databases.
PASS application is useful because it gives the hits in the following:
Revealing new effects and mechanisms of action for the old substances in corporate and private data bases.
Finding the most probable new leads with required activity spectra among the compounds from in-house and commercial databases.
Selecting the most prospective compounds for high throughput screening from the set of available samples.
Determining the screens that are more relevant for a particular compound.
Revealing New Effects and Mechanisms of Action is considered below on the example of predicting the biological activity spectrum for well-known cerebrotonic drug Cavinton (Vinpocetin) launched by Gedeon Richter (Hungary) more than twenty years ago. Its structural formula and predicted biological activity spectrum are given below.
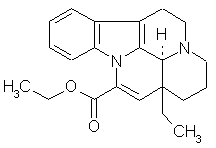
Predicted biological activity spectrum for Cavinton
45 Descriptors, 0 New Descriptors, 47 Predicted Activities
No |
Pa |
Pi |
Activity |
Experiment |
Reference |
1 |
0.929 |
0.004 |
Peripheral vasodilator | ||
2 |
0.900 |
0.000 |
Multiple sclerosis treatment | ||
3 |
0.855 |
0.005 |
Vasodilator | + |
|
4 |
0.844 |
0.003 |
Abortion inducer | + |
[17] |
5 |
0.812 |
0.001 |
Antineoplastic enhancer | ||
6 |
0.760 |
0.006 |
Coronary vasodilator | + |
[19] |
7 |
0.732 |
0.007 |
Spasmogenic | ||
8 |
0.700 |
0.036 |
Antihypoxic | + |
|
9 |
0.650 |
0.004 |
Lipid peroxidase inhibitor | + |
|
10 |
0.648 |
0.008 |
Cognition disorders treatment | + |
|
11 |
0.656 |
0.021 |
Antiischemic | + |
|
12 |
0.577 |
0.013 |
Acute neurologic disorders treatment | + |
|
13 |
0.540 |
0.039 |
Spasmolytic | + |
[18] |
14 |
0.519 |
0.026 |
Antianginal agent | ||
15 |
0.486 |
0.037 |
Antihypertensive | + |
[18] |
16 |
0.449 |
0.035 |
Antiarrhythmic | + |
[29] |
17 |
0.432 |
0.063 |
Sympatholytic | ||
18 |
0.438 |
0.077 |
Sedative | + |
[18] |
19 |
0.500 |
0.152 |
Antiinflammatory, Pancreatic | ||
20 |
0.328 |
0.020 |
Antidepressant, Imipramin-like | ||
21 |
0.300 |
0.010 |
Thrombolytic | + |
|
22 |
0.342 |
0.075 |
Psychotropic | + |
[18] |
23 |
0.276 |
0.023 |
Alpha 2 adrenoreceptor antagonist | + |
[30] |
24 |
0.273 |
0.029 |
Anesthetic intravenous | ||
25 |
0.547 |
0.304 |
Vascular (periferal) disease treatment | ||
26 |
0.225 |
0.006 |
Antineoplastic Alkaloid | ||
27 |
0.291 |
0.086 |
Cholinergic antagonist | ||
28 |
0.263 |
0.066 |
Benzodiazepine agonist partial | ||
29 |
0.417 |
0.238 |
Insulin promoter | ||
30 |
0.222 |
0.045 |
MAO-A inhibitor | ||
31 |
0.353 |
0.188 |
Cardiovascular analeptic | ||
32 |
0.249 |
0.100 |
Narcotic antagonist | ||
33 |
0.300 |
0.161 |
Acetylcholine release stimulant | ||
34 |
0.236 |
0.104 |
Antitumor-cytostatic | ||
35 |
0.271 |
0.165 |
Antiparkinsonian, rigidity relieving | ||
36 |
0.218 |
0.127 |
Antidepressant | ||
37 |
0.247 |
0.157 |
Analeptic | ||
38 |
0.211 |
0.126 |
Potassium channel antagonist | ||
39 |
0.243 |
0.158 |
Antiparkinsonian, tremor relieving | ||
40 |
0.333 |
0.258 |
5 Hydroxytryptamine 3 agonist | ||
41 |
0.233 |
0.172 |
Respiratory analeptic | ||
42 |
0.242 |
0.184 |
Antipsoriatic | ||
43 |
0.131 |
0.081 |
Analgesic, opioid | ||
44 |
0.147 |
0.128 |
N-cholinergic agonist | ||
45 |
0.285 |
0.267 |
cAMP phosphodiesterase inhibitor | + |
[17] |
46 |
0.175 |
0.162 |
Anestetic general | ||
47 |
0.375 |
0.370 |
Male reproductive disfunction treatment |
As Cavinton is used in medicinal practice for twenty years, many activities that were found in pre-clinical testing and clinical trials during this period are compared with the result of prediction. According to the available literature only 16 of 47 predicted activities of Cavinton are already found. These activities are marked by "+" in the Table above.
In particular, computer system PASS predicts the vasodilator and spasmolytic activities (Pa=0.855 and 0.540 respectively). It corresponds to well-known pharmacological effects of Cavinton, which causes the vasodilatation, increases the brain blood flow and metabolism. Antihypoxic and Antiischemic actions are also predicted for Cavinton (Pa=0.700 and 0.656 respectively). Really, Cavinon is used for these purposes. Cavinton is predicted as Lipid peroxidase inhibitor (Pa=0.650), Agent for cognition disorders treatment (0.648), Agent for acute neurological disorders treatment (0.577), etc. Cavinton has all these activities.
In predicted biological activity spectrum of Cavinton there are several actions which might become the basis for new application of the substance. Among them: Multiple sclerosis treatment (Pa=0.900); Antineoplastic enhancer (0.812), Antineoplastic Alkaloid (0.225) and Antitumor-Cytostatic (0.236); Antiparkinsonian rigidity-relieving (0.271) and Antiparkinsonian tremor-relieving (0.243); etc. While the Multiple sclerosis treatment is predicted with high probability, all other additionally predicted activities have a relatively small values of Pa. Thus, if these actions will be confirmed in the experiment, it might be the discovery of New Chemical Entities (NCE).
Similarly, the predicted activity spectrum for any compound provides the basis for its further testing. As a result some new effects and mechanisms can be found for old substances. Varying the cutoff value of Pa one may choose the desirable level of novelty vs. acceptable risk of negative result.
Finding the Most Probable New Leads with Required Activity Spectra. If the researcher can define which activities are desirable and which are not desirable for a compound according to the List of Activities (http://www.ibmh.msk.su/PASS/descript33.htm) predicted by PASS, she can select such compounds from the set of structures, which are available from in-house and commercial databases. For example, among the 15630 compounds from database of samples available in stock of ChemStar (http://www.chemstar-ru.com) for which PASS prediction was carried out, 959 compounds are predicted as Endothelin antagonist, 236 compounds as Angiotensin II antagonist, 57 compounds as Angiotensin converting enzyme inhibitor. If the purpose of the study is to find the compounds with dual mechanism of Antihypertensive effect, e.g. Angiotensin converting enzyme inhibitor + Endothelin antagonist, only 11 compounds are predicted as having both activities. The best from the hits has Pa =0.170 (Endothelin antagonist) and Pa=0.244 (Angiotensin converting enzyme inhibitor). Based on this result one may decide either to test these 11 compounds or to carry out the prediction and selection for compounds from another database. In any case varying the cutoff value of Pa it is possible to choose the compounds with less or higher novelty (see: Interpretation of the Prediction's Results).
Selecting the Most Prospective Compounds for Highthroughput Screening. If the searched leads should have the activities which are included into the list of 408 ones, predicted by PASS-C 4.00, probably, the strategy considered in previous section is the best. However, sometimes either the pharmacological target for which leads are searched is rather new and there are no compounds in the PASS training set related to this activity, or the Company would not like to disclose its fields of interests. In such case two other strategies are suitable.
The first strategy is based on suggestion that the more kinds of activity are predicted as probable for a compound, the more probable to find any useful pharmacological action in it. For each compound from available set of samples the following value is calculated:
P = [S Pa/(Pa+Pi )]/n
where n is the number of biological activities under consideration (in PASS-C 4.00 n = 408).
All compounds are arranged in the descending order of P values, and only compounds with the highest values of P which have the highest biological "potential" are selected for screening.
The second strategy is based on suggestion that the more is "novelty" of compounds relating to the compounds from the training set of PASS, the higher is probability to find NCE. Thus, the compounds with the highest amount of new descriptors have to be included into this sub-set.
Both strategies were tested on the datasets included 10,000 - 70,000 compounds and their efficacy is shown [31].
Determining the Screens that are More Relevant for a Particular Compound. Based on the predicted activity spectrum for new compound, its testing can be organized in descending order of difference (Pa-Pi) for different activities. For example, if we consider the given above example of Cavinton, it should be studied in the following tests: Peripheral vasodilator (0.929-0.004), Multiple sclerosis treatment (0.900-0.000), Vasodilator (0.855-0.005), Abortion inducer (0.844-0.003), Antineoplastic enhancer (0.812-0.001), Coronary vasodilator (0.760-0.006), etc.
In this case both safety and efficacy of new compound will be characterized in more comprehensive way. Moreover, it is shown that the economic viability of such approach to testing is more than 500% [32]. Certainly, in a particular case the researcher will take into account also her facility of testing.
PASS value is proved by many different compounds from various chemical series for which the result of computer prediction is confirmed by experiment. Some of these examples are given below.
The activity spectra have been predicted for 300 new chemical compounds, synthesized in the Chemical-Pharmaceutical Research Institute (Novokuznetzk). Twenty compounds have been selected for testing as probable antiulcer agents. Nine compounds have been synthesized and tested. The potent antiulzer activity is found for 5 of these compounds. These new antiulcer agents are NCE [33]. The economic viability is about (300/20)100 = 1500% in this study.
The activity spectra have been predicted for 520 new chemical compounds, synthesized in the Institute of Organic Chemistry of Russian Academy of Science (Moscow). Fourteen compounds have been selected for testing as the most prospective. It is shown that the results of 22 experiments made on 5 various kinds of activity, coincide with predictions in 20 cases. The accuracy of prediction is about 90%.
Based on the predicted biological activity spectra for about 20 macroheterocyclic compounds, 2 antitumor leads were found among them [34].
New antibacterial agents were found based on prediction of biological activity spectra for derivatives of 1-amino-4-(5-arylozaxolyl-2)-butadiens-1,3 [35].
Analgesic, antiinflammatory, antioxidant and some additional activities were predicted and confirmed by experiment for some thiazole derivatives [36].
These and some other examples demonstrate clearly that the approach to predicting many biological activities simultaneously can be effectively applied to compounds from different chemical series to find various pharmacological actions.
Naturally, the PASS approach has some limitations. They are:
PASS approach can be applied to so-called "drug-like" substances.
PASS approach can be applied to the activities for which the training set will include no less than 5 active compounds per activity.
The accuracy of PASS prediction is significantly more than in random guess-work but still limited. For essentially new compounds that have no one descriptor occurred in the training set PASS cannot predict the activity spectrum at all.
In some cases PASS predicts as probable both agonist's and antagonist's (blocker and stimulator) actions simultaneously. Thus, only experiments can clarify the intrinsic activity of a compound, but it probably has an affinity to appropriate receptor (enzyme).
PASS does not predict if the compound will become a drug, but helps to select the most prospective leads.
Since July 1998 PASS-C 4.00 is open for free testing via Internet (http://www.ibmh.msk.su/PASS/default.htm). Anyone who would like to obtain the additional information about biological potential of her chemical compound may fill the registration form and send the structure file in ISIS (MDL Information Systems, Inc.) "mol" format.
Such file can be prepared, for example, with chemical editor ISIS/Draw (MDL Information Systems, Inc.). ISIS/Draw is available free for personal or non-commercial use from the MDL web site http://www.mdli.com.
The molfile have to be prepared with ISIS/Draw in the following way. Structure is drawn on the display using the options of menu and the mouse. After that one can choose "Edit" a "Select All". When the molecule is selected as a total, choose "File" a "Export" a "Molfile". File have to be saved on the disk under the particular name defined by the user.
When the molfile is prepared and the registration form is once filled in the Internet version of PASS, one may click on the option "Browse" and select the molfile name on her disk and click on "Open". This name have to be appeared in the window near the option "Browse". After that she has to click on "Submit now" and wait for result on her display. In case of any problem, it can be solved by E-mail: pass@ibmh.msk.su.
New computer approach to predicting biological activity for a drug-like compound on the basis of its structural formula is developed. Its applicability and effectivity in finding new leads is demonstrated on examples of both compounds with known activities and new synthesized structures studied as potential pharmacological agents. Easy access via Internet provides the feasibility for its testing and use by any researcher or institution.
We are gratefully acknowledge MDL Information Systems, Inc. for providing the ISIS/Host, ISIS/Base and MDDR database which were used in this study.
FLUOROURACIL'S KNOWN ACTIVITIES (CAS NO. 51-21-8)
| Activity | Publ. Year |
| Antineoplastic | 1962 |
| Embryotoxic | 1966 |
| Antiviral | 1971 |
| Thimidine Triphosphate Synthesis Inhibition | 1977 |
| RNA Synthesis Inhibition | 1980 |
| Protein Synthesis Inhibition | 1987 |
| Lipid Metabolism Regulator | 1990 |
| Immunosuppressant | 1990 |
| Antimetabolite | 1991 |
| Antiviral (AIDS) | 1996 |
. . . ? |
. . . ? |
EXAMPLES OF PHARMACEUTICALS, WHICH ADDITIONALLY DISCOVERED
ACTIVITY
WAS USED FOR ANOTHER INDICATION
| Pharmaceutical | Therapeutic Effect | Year |
| Acetazolamide | Diuretic
Antiepileptic |
1954 1956 |
| Valproate | Anxiolytic
Antiepileptic |
1961 1989 |
| Levamisole | Antihelmintic
Immunostimulant |
1968 1980 |
| Alprostadil | Antiagregant
Erectant |
1988 1994 |
| Aspirin | Analgesic
Antiagregant |
1899 1971 |
? |
? |
? |
Prous J. Drug Years News-1995.
[1] Wermuth C.G., ed., Medicinal chemistry in practice, Academic Press, London, 1996, 968 p.p.
[2] Van de Waterbeemd H., ed., Structure-property correlations in drug research, Landes, Austin, 1996, 210 p.p.
[3] Dean P.M., Molecular similarity in drug design, Blackie Academic, London, 1995,
[4] Livingstone D., Data analysis for chemists. Applications to QSAR and Chemical Product Design, Oxford Science Publ., Oxford, 1995, 239 p.p.
[5] Kubinyi H., ed., 3D QSAR in drug design, Escom, Leiden, 1993, 759 p.p.
[6] Avidon V., Criteria for similarity assessment of chemical structures and the basics of informational language for development of informational-logical system on biologically active compounds. Chem. & Pharmaceut. J. (Rus.), 1974, 8 (8), 22-25.
[7] Piruzyan L.A., Avidon V.V., Rozenblit A.B., et.al. Statistical analysis of the information file on biologically active compounds. I. Data base on the structure and activity of biologically active compounds. Chem. & Pharmaceut. J. (Rus.), 1977, 11 (4), 35-40.
[8] Piruzyan L.A., Rudzit E.A. The methodical approaches to study biological activity of chemical compounds. Chem. & Pharmaceut. J. (Rus.), 1976, 10 (8), 21-27.
[9] Burov Yu.V., Korolchenko L.V., Poroikov V.V. National system for registration and biological testing of chemical compounds: facilities for new drugs' search. Bull. Natl. Center for Biologically Active Compounds (Rus.), 1990, No. 1, 4-25.
[10] Filimonov D.A., Poroikov V.V., Karaicheva E.I., et. al. (1995). Computer-aided prediction of biological activity spectra of chemical substances on the basis of their structural formulae: computerized system PASS. Experimental and Clinical Pharmacology (Rus), 58 (2), 56-62.
[11] Filimonov D.A., Poroikov V.V. PASS: Computerized prediction of biological activity spectra for chemical substances. Bioactive Compound Design: Possibilities for Industrial Use, BIOS Scientific Publishers, Oxford, 1996, p.47-56.
[12] Poroikov V.V., Filimonov D.A. Computerized prediction of biological activity spectra for chemical substance - new approach to effective drug design. In: QSAR and Molecular Modelling Concepts, Computational Tools and Biological Applications. Barcelona: Prous Science Publishers, 1996, p.49-50.
[13] Poroikov V.V., Filimonov D.A., Stepanchikova A.V., et.al.. Opimization of synthesis and pharmacological testing of new compounds based on computerized prediction of their biological activity spectra. Chem. & Pharmaceut. J. (Rus), 1996, 30 (9), 20-23. (English translation by Consultants Bureau, New York: Pharmaceutical Chemistry Journal, 1996, 30 (9), 570-573).
[14] Poroikov V.V. PASS, a program for the prediction of activity spectra from molecular structure. Newsletter of The QSAR and Modelling Society, 1997, No. 8, 12-15.
[15] Gloriozova T.A., Filimonov D.A., Lagunin A.A., Poroikov V.V. Testing of computer system for prediction of biological activity spectra PASS on the set of new chemical compounds. Chem. & Pharmaceut. J. (Rus), 1996, In press.
[16] Filimonov D.A. Comparison of Algorithms for Computer Prediction of Biological Activity Spectra for Chemical Compounds on the Basis of Their Structural Formulae. II Rus. Natl. Congress "Man and Drugs", Moscow, Abstracts, 1995, 62-63.
[17] Summary of Cavinton (Vinpocetine) Gedeon Richter, Budapest-Hungary, 1994-06-07.
[18] Mashkovskii M.D. The Pharmaceuticals, Medicine, Moscow, 1997, v.1, 399-400.
[19] VIDAL. Pharmaceuticals in Russia. Moscow, AstraPharmService, 1997.
[20] Kiss B., Karpati E. Acta Pharm. Hung., 1996, 66 (5), 213-224.
[21] Plotnikova T.M., Plotnikov M.V., Bazhenova T.G. Bull. Exp. Biol. Med., 1991, 111 (2), 170-172.
[22] Karmazsin L., Olah V. A., Balla G., Makay A. Acta Paediatr. Hung. 1990, 30 (2), 217-224.
[23] Suno M., Nagaoka A. Nippon Yakurigaku Zasshi, 1988, 91 (5), 295-299.
[24] Boda J., Karsay K., Czako L., Fugi S., Kovacs A., Koncz I., Maczko P. A. Ther. Hung., 1989, 37 (3), 176-180.
[25] Molnar P., Gaal L. Eur. J. Pharmacol., 1992, 215 (1), 17-22.
[26] Kiss B., Karpati E. Acta Pharm. Hung., 1996, 66 (5), 213-224.
[27] Hadjiev D., Yancheva S. Arzneimittelforschung, 1976, 26 (10A), 1947-1950.
[28] Rischke R., Krieglstein J. Pharmacology, 1990, 41 (3), 153-160.
[29] Karpati E., Szporny L. Arzneimittelforschung, 1976, 26 (10A),1908-1912.
[30] Paulo T., Toth P.T., Nguyen T.T., Forgacs L., Torok T.L., Magyar K. J. Pharm. Pharmacol., 1986, 38 (9), 668-73.
[31] Poroikov V.V., Filimonov D.A., Stepanchikova A.V. Biological Activity Spectra Prediction as a Tool to Select the Most Prospective Compounds from Commercial and In-House Databases. Abstr. Intern. Med. Chem. Symp., Seoul, 1997, P.143.
[32] Poroikov V.V, Filimonov D.A, Boudunova A.P. Computer Assisted Prediction of Biological Activity Spectra: Estimating the Effectivity of Use in High Throughput Screening. Abstr: XIVth International Symposium on Medicinal Chemistry, Maastricht, the Netherlands, 1996, P-3.05.
[33] Trapkov V.A., Budunova A.P., Burova O.A., Filimonov D.A., Poroikov V.V. Discovery of New Antiulcer Agents by Computer Aided Prediction of Biological Activity. Problems in Medical Chemistry (Moscow), 1997, 43 (1), 41-57.
[34] Islyaikin M.K., Danilova E.A., Kudrik E.V., Smirnov R.P., Boudunova A.P., Kinzirskii A.S. Synthesis and study of antitumor action of macroheterocyclic compounds and their complexes with metals. Chemical & Pharmaceutical J. (Rus), 1997, 31 (8), 19-22.
[35] Maiboroda D.A., Babaev E.V., Goncharenko L.V. (1998). Synthesis and study of spectral and pharmacological properties of 1-amino-4-(5-arylozaxolyl-2)-butadiens-1,3. Chemical & Pharmaceutical J. (Rus), 32 (6), 24-28.
[36] Geronikaki A., Poroikov V., Hajipavlou-Litina D., Mgonzo R., Filimonov D., Lagunin A. Synthesis, computer assisted prediction of biological activity spectra and experimental testing of new thiazole derivatives. Quantitative Structure-Activity Relationships, 1998, In press